Activation of carbon dioxide by electrochemical reduction of molybdenum hexacarbonyl in aprotic solvent: a combined IR spectroelectrochemical and DFT calculation study.
F. Gloaguen, N. Le Poul, Transition Metal Chemistry 2025, DOI: 10.1007/s11243-025-00659-1
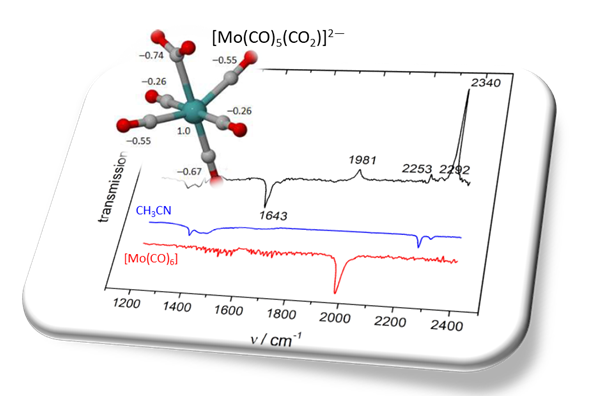
The mechanism of carbon dioxide (CO2) activation by the electrochemical reduction of molybdenum hexacarbonyl (Mo(CO)6) in dry organic solvent was reinvestigated using IR spectroelectrochemistry (IR-SEC) combined with density functional theory (DFT) calculations. Cyclic voltammetry (CV) and IR-SEC experiments, carried out under inert atmosphere, confirmed that the stable pentacarbonyl dianion [Mo(CO)5]2− is readily formed at the reduction potential of the hexacarbonyl parent complex. In addition, IR-SEC monitoring of the reduction of Mo(CO)6 in CO2-saturated solution showed an absorption band ascribed to the formation of bicarbonate (HCO3−), but no signs for the formation of formate (HCO2−) or oxalate (C2O42−). These experimental results were rationalized by DFT calculations on the coordination mode of CO2 to [Mo(CO)5]2−. Indeed, no stable structure could be calculated for the η1-OCO isomer, whereas the optimized structure of the η2-CO2 isomer was calculated to be energetically less stable than that of the η1-CO2 isomer, the latter being identified as a key intermediate for the selective formation of carbon monoxide (CO) and water (H2O) upon O-protonation of the CO2-adduct. This catalytic behavior is discussed here in terms of Mulliken atomic charge redistribution over the CO2 binding and activation processes, and compared with what was previously reported for tetracarbonyl Mo-diimine complexes, where diimine ligands display “redox non-innocent” properties.